IRANDERMA |
|
Quiz: March 2005 |
An 8-year-old girl presented with sevreal months history for facial rash. Physical examinations revealed also scaly papules on the knuckles and knees. ANA was negative... What is your diagnosis?
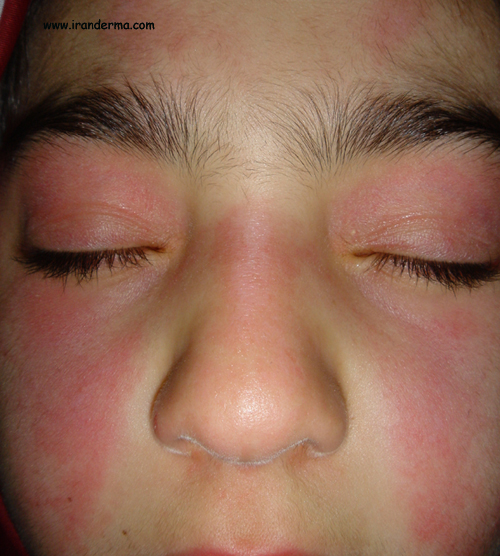
Diagnosis: Juvenile Dermatomyositis
By: Mehrdad Mehravaran, M.D., Szeged-Hungary
Dermatomyositis-Ploymyositis
(DM-PM) are inflmmatory myopathies, which have prominent cutaneous
findings. They may be primary or may be associated with connective tissue
diseases, they have not been conclusively shown to have an association
with malignacy. Childhood DM-PM are distinct entity from adulds DM-PM.
Children or juvenile DM-PM are more likely than adults to have extensive
calconosis and vasculitis.
DM_PM are inflammatory diseases
primarly affecting striated muscles, estimated incidence about 1/200,000
per year, females are affected more than males and are more frequent in
black women.
Porposed classification by Bohan
et al 1
as
follows:
- group 1 primary idiopathic polymysitis (the
most common type)
- group 2 primary idiopathic dermatomysitis
- group 3 dermatomysitis or polymysitis
associated with neoplasia
- group 4 childhood dermatitis or polymysitis
- group 5 polymysitis or dermatomysitis with
associated connective tissue disease (overlap syndromes)
Diagnosis:
The five major diagnostic
criteria:
- progressive symmetrical proximal muscle
weakness
- abnormal
muscle biopsy consistent with polymysitis
- elevation of muscle enzymes in the serum
(e.g., creatine phosphokinase and aldolase
- abnormal electromygram, consistent with
polymysitis
- characterisctic rash od dermatomysitis
The diagnosis of PM is
established if patients have all four noncutaneous criteria; it is
probable if three criteria are present and possible if two are present.
The diagnosis of DM is established if four criteria (including the
cutaneous criterion) are present and probable if three are present.
Pathologic
and labratory features:
The cardinal pathologic features
of DM-PM are abnormal muscle enzymes and abnormal findings on muscle
biopsy and on electromyography.
Muscle
enzymes
are the most common sensitive indicators of disease activity, and
cratinine phosphokinase (CPK) and aldolase are more sensitive and
informative than transaminases or lactic dehydrogenase. Although none of
these enzymes is specific for striated muscle injury, CPK isoenzyme levels
may be used to distinguish stirated from smooth muscle injury and CPK
levels can be useful in monitoring the course of the disease and the
response to therapy. Serum
enzymes levels are elvated in over 95% of patients with active DM-PM but
may be normal in those who have inactive disease or extensive loss of the
muscle mass.
Muscle
biopsy
is abnormal in about 90% of patients. Since findings may be focal,
abnormalities can be missed on a single biopsy. Biopsies may show a
segmental necrosis of muscle fibers, an inflammatory infiltrate, and focal
areas of muscle regeneration. The inflammatory infiltrate consists of
variable amounts of macrophages, with phagocytosis of degenerating muscle
fibers and a patchy infiltrate of lymphocytes and a few plasma cells. In
children vascular injury isprominent.
Electromyography
(EMG)
shows abnormalites in about 80% of patient. It is useful in confirming the
diagnosis of myopathy and in providing evidence against neuropathy.
Abnormalites include low-amlitude, short-duration, and polyphasic
potentials; spontaneous fibrillation; increased irritability on insertion
of electrodes; and positiv sharp waves.
Skin
histopathology is nondiagnostic; often, findings are only those
of nonspecific inflammation.
DM-PM constitute a group of
autoimmune diseases in which autoantibodies
are often apparently absent, with 60% or fewer patients reported to have
abnormal test for antinuclear antibodies (ANA). Several autoantibodies
described in DM-PM are apparently specific for these diseases but are not
detected in the majority of patients. These include antibodies to histidyl
tRNA synthetase (Jo-1), which are found in about 25% of PM patients, and
antibodies to threonyl tRNA synthetase (PL-7) and alanyl tRNA
synthetase (PL-12), which are found in a small fraction of patients.
Anti-Jo-1 antibodies are found
more often in PM than in DM patients and appear to be most common in
patients with PM who have interstitial lung disease. Antibodies to the
Mi-2 antigen appear to be relatively specific for DM but are found in a
minority of patients. Antibodies
to a complex of proteins called PM-Scl are found in patients with
PM-scleroderma overlap syndrome.
Cutaneous
lesions
The classic eruption of DM is
erythema involving the face, a reddish purple color being prominent of
eyelids. The rash in DM can precede clinical indications of muscle
weakness, generally by a few weeks or months, but typical cutaneous
lesions can appear as much as 4 years prior to clinically evident
myopathy, and there are rare patients who never develope demostrable
muscle diseases. The eruption of the eyelids is often called a helitrope rash.
The facial erythema in DM is
most apparent on the malar areas and eyelids and may be indistinguishable
from the lesions of acute cutaneous lupus erythematosus. Scaly, erythematous lesions can involve not only in face but
also in the neck, upper trunk, and extensor exterimites (eg, elbows and
knees). Photosensitivity
is common in DM.
Characteristic lesions occur on
the hands and may be very helpful in distinguishing DM from LE. Erythema
in DM occurs over the knuckles rather than over the phalanges as in lupus.
Violaceous erythema, with or without scale, over the knuckles,
elbows, knees, and medial malleoli is known as Gottron’s
sign. In addition to erythema, raised violaceous, flat topped
lesions, called Gottron’s papules, may be present on the knunckles.
Othe skin signs are periungual
erythema, telangiectasias, fragmentation of the cutiles, nail fold
capillary abnormalities, poikilodermatous skin changes (the affcted area
consists of widespread, mottled hyper- and hypopigmentation,
withtelangiectasias and atrophoy and sometimes with ulcerations),
cutaneous calcification (are typical in childhood DM), Raynaud’s
phenomenon and sclerodactly.
Internal
Oragan Involvement
Systemic
findings:
The onset of systemic problems
is usually insidious, beginning with fever,
malaise and diffuz weakness. Proximal muscle weakness is present in virtually all patients.
Patients often experiance diffliculty in climbing stairs or rising from
the floor or from a low chair. Weakness may progress to involve the
shoulder gridle, and patients may have difficulty in raising the arms
above the head, as when combing their hair. Neck muscles, facial muscles,
and pharyngeal muscles may be affected, and muscle pain and tenderness may
occur. Involvement of the appropriate muscles can lead to dysphagia,
dyspnea or dysphonia. Reflexes usually present but reduced,
sensory disturbances do not occur.
Cardiac
involvement
consiting of myocarditis and/or conduction system abnormality is seen in
most patients with adult DM-PM. Lung disease is also very common and can be a result of
interstitial fibrosis, aspiration penumonia due to weakened pharyngeal
muscles, or hyperventilation due to involvement of the respiratory
muscles. Esophageal dysmotility
is a frequent finding. Cervical dysphagia
and nasopharyngeal reflux
are seen in DM-PM. Arthritis
is seen occationally in patients with the primary disorders and often in
those with an overlap syndrome.
Marked muscle destruction can
lead to myoglobinurea and acute renal failure. Dysphagia can complicate
the pulmonary picture and causing aspiration penumonia.
In children acute
gasterointestinal problems may develop secondary to necrotizing arteritis
with ulcerations and infarctions. Other difficulties include ocular muscle
weakness, carpal tunnel syndrome, osteoprosis secondary to
corticosteroids.
In fulminant cases, such as
tumor associated dermatomyositis is adult the patient may wind up
virtually helpless. In children such dramatic courses are uncommon.
Whiles arthiritis or Raynoaud
phenomenon suggest an overlap syndrome, arthralgias and morning stiffness
are often present. Arthritis may also associated with pulmonary fibrosis
and anti-Joi antibodies (in children pulmonary involvement is uncommon).
Childhood
(Juvenile) DM-PM,
vasculitis is much more common than it is in the adult forms. The
gastrointestinal tract is often affected, with resultant ulceration,
bleeding, and perforation. Calcinosis is more common in children and can
involve muscle and tendon as well as skin.
Malignancies
of various internal organs have been reported to be associated with DM-PM,
but it is still nor clear if this is a true relationship is a subgroup of
adult patients. Juvenile DM is not associated with malignancy.
Juvenile
DM
Both childhood and adult forms
of DM and PM adhere to same diagnostic criteria, but childhood disease
have unique features of vasculitis and calcinosis. Juvenile DM have two
presentations. Approximately one half of the patients have rapidly
progressive diseases with a high mortalityrate characterized by fever,
anorexia, and clinical and histologic vasculitis in addition to classic
cutanepus and muscle abnormalities. Vasculitis can result in GI hemorrhage
and even bowel perforation. Therapeutic response and prognosis are poor.
The remainder of children have a more subacute presentation with gradual
progression of muscle weakness and subsequent calcinosis. Clacinosis can
be superficial; subcutaneous near joints (knee, elbows, fingers); in the
fascial planes within muscle; or rarely as a diffuse exoskeleton.
Managment
and treatment:
- The mainstay of therapy is sytemic steroids
with relatively high dose (1mg/kg/day) until improvement is seen and
then tapered very slowly, often by no more than 10% per month.
- In refractory cases if the response to
steroids is insufficient, cytostatic drugs may be added. Mehtotrexate,
cyclophasphamide, chlorambucil, azathioprine, plasmapheresis, and more
recently Intravenous Immunoglobin (IVIgs) are advocated.
- Cutaneous lesions can be treated with topical
corticosteroids and antimalatial agents.
References:
- Bohan A, peter JB: polymysitis and
dermatomysitis. N Engl J Med 1975;292:344-347, 403-407.
![]()
ايران درما |